Sobre Progeria
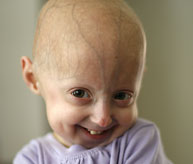 Síndrome de Progeria de Hutchinson-Gilford (“Progeria” ou “HGPS”) é uma condição genética rara e fatal caracterizada por uma aparência de envelhecimento acelerado em crianças. Seu nome é derivado do grego e significa “prematuramente velho”. Embora existam diferentes formas de Progeria*, o tipo clássico é a Síndrome de Progeria de Hutchinson-Gilford, que recebeu o nome dos médicos que a descreveram pela primeira vez na Inglaterra; em 1886 pelo Dr. Jonathan Hutchinson e em 1897 pelo Dr. Hastings Gilford.
Síndrome de Progeria de Hutchinson-Gilford (“Progeria” ou “HGPS”) é uma condição genética rara e fatal caracterizada por uma aparência de envelhecimento acelerado em crianças. Seu nome é derivado do grego e significa “prematuramente velho”. Embora existam diferentes formas de Progeria*, o tipo clássico é a Síndrome de Progeria de Hutchinson-Gilford, que recebeu o nome dos médicos que a descreveram pela primeira vez na Inglaterra; em 1886 pelo Dr. Jonathan Hutchinson e em 1897 pelo Dr. Hastings Gilford.
A HGPS é causada por uma mutação no gene chamado LMNA (pronuncia-se lamin – a). O gene LMNA produz a proteína Lamin A, que é o andaime estrutural que mantém o núcleo de uma célula unido. Os pesquisadores agora acreditam que a proteína Lamin A defeituosa torna o núcleo instável. Essa instabilidade celular parece levar ao processo de envelhecimento prematuro na Progéria.
Embora nasçam parecendo saudáveis, crianças com Progeria começam a apresentar muitas características de envelhecimento acelerado nos primeiros dois anos de vida. Os sinais de Progeria incluem falha de crescimento, perda de gordura corporal e cabelo, pele com aparência envelhecida, rigidez das articulações, luxação do quadril, aterosclerose generalizada, doença cardiovascular (coração) e derrame. As crianças têm uma aparência notavelmente semelhante, apesar das diferentes origens étnicas. Sem tratamento, crianças com Progeria morrem de aterosclerose (doença cardíaca) com uma idade média de 14,5 anos.
* Outras síndromes progeroides incluem a síndrome de Werner, também conhecida como “progéria adulta”, que não começa até o final da adolescência, com uma expectativa de vida de até os 40 e 50 anos.
Inscrever-se
para o nosso
Atualizações!
Juntos, nós
VAI
encontre a cura!
